Universal MLPs (UMLIPs)
Universal MLPs (UMLIPs) are a class of MLPs that are designed to be transferable across different systems and chemical environments. They are trained on a large and diverse dataset of atomic configurations and their corresponding DFT energies, forces, and stresses. The goal of universal MLPs is to provide a single model that can accurately predict the potential energy surface for a wide range of materials and systems without the need for retraining or fine-tuning.
We have used MACE-MP0 in our previous lectures and assignment.
Training Dataset¶
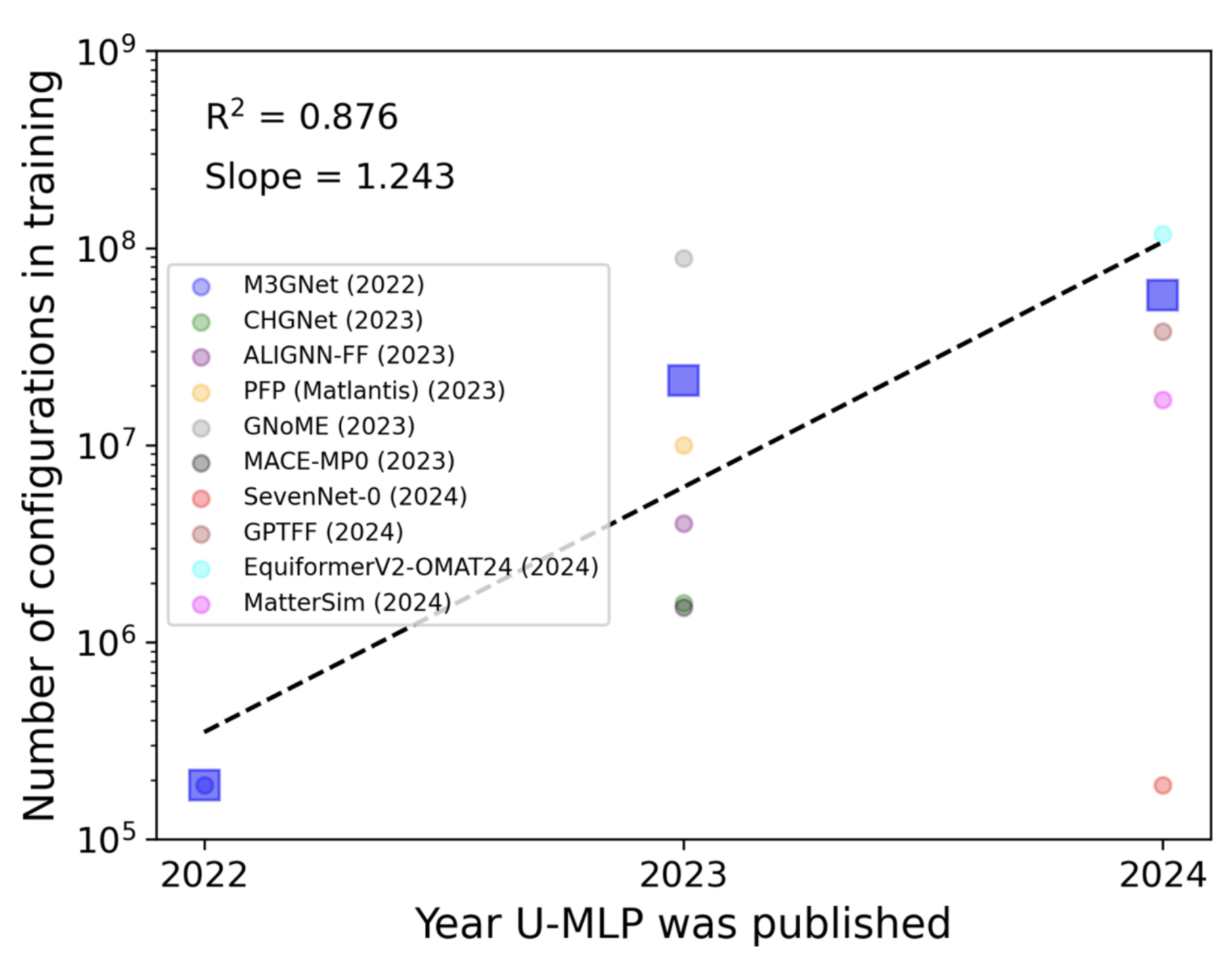
Dataset size of the training data used in UMLPs. The size of the dataset keeps increasing. The average size of the training dataset can be 108 atomic configurations in 2024.
Typical UMLPs cover 10-100 elements and trained using large dataset such as the MPtrj, OMAT24, and Alexandria. They are summarized in the table below (taken from here)
Examples¶
| U-MLIP Name | Training Database | Number of Elements | Training Data | Notes |
|---|---|---|---|---|
| M3GNet | Materials Project | 89 | 62,783 compounds: 187,687 energies, 16,875,138 forces, and 1,689,183 stresses | Training data taken from Materials Project dating back to its inception in 2011 |
| CHGNet | Materials Project + Trajectory database | 89 | 146,000 compounds: 1,580,395 energies, 49,295,660 forces, and 14,223,555 stresses | Training data taken from Materials Project GGA and GGA + U relaxation trajectory up to Sept 2022 |
| ALIGNN-FF | JARVIS-DFT | 89 | 307,113 energies and 3,197,795 forces for 72,708 compounds | |
| PFP (Matlantis) | Custom | 96 | Roughly 60 million configurations | Training data is a custom in-house set performed by a collaboration of Preferred Networks, Inc. and ENEOS Corporation |
| GNoME | Materials Project + Custom | 94 | Roughly 89 million configurations from 6 million compositions | Initial training done on Materials Project data from 2018 comprising 69,000 materials. Later fits include about 89 million configurations |
| MACE-MP0 | Materials Project + Trajectory database | 89 | Same training data as used to build the CHGNet potential | An additional dispersion correction model can be used to accurately capture dispersion physics not present in the training data |
| SevenNet-0 | Materials Project | 89 | Same training data as used to build the CHGNet potential | Same training data as used to build the M3GNet potential |
| GPTFF | Atomly.net | Value not given in text | Roughly 2.2 million crystal structures, consisting of a total of 37.8 million energies (349 k of these are equilibrium states), 11.7 billion force vectors, and 340.2 million stresses | |
| MatterSim | Initial data from public databases like Materials Project, Materials Project Trajectory, and Alexandria, then customized with additional DFT calculations | 89 | Roughly 17 million atomic configurations | Sampling techniques include simulations with temperatures ranging from 0-5000 K and pressures from 0 to 1000 GPa |
| Orb | Materials Project Trajectory and Alexandria | 89 | Value not directly mentioned in text | |
| EquiformerV2OMAT24 | Initial data from public databases like Materials Project, Materials Project Trajectory, and Alexandria, then customized with additional DFT calculations | 89 | Roughly 118 million atomic configurations | As of this writing, state-of-the-art performance on the main ranking of the MatBench leaderboard and largest publicly-available DFT database |
Benchmarking UMLPs¶
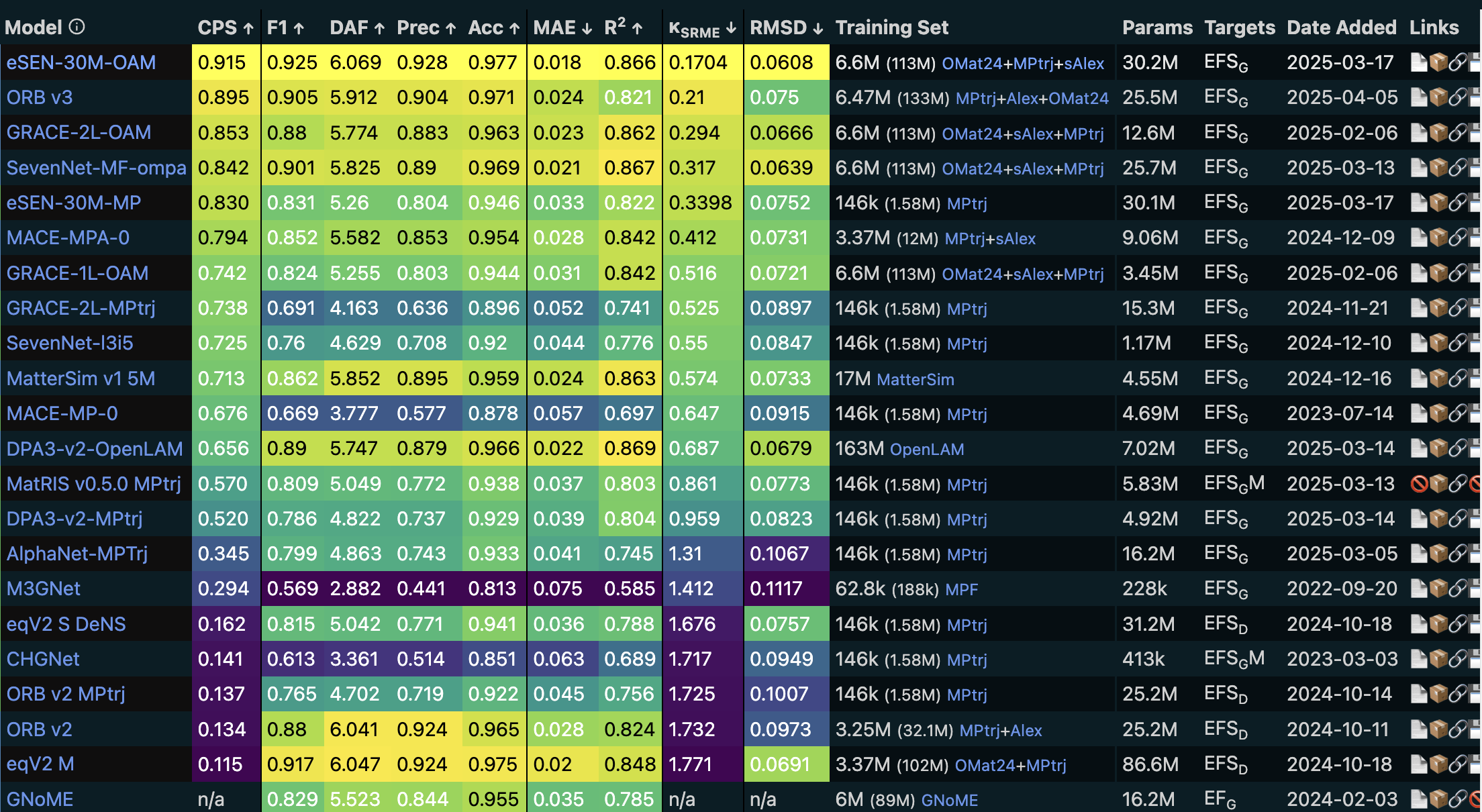
The Matbench Discovery benchmark table for various UMLPs., data accessed on 2025-04-16.
There are established benchmarks like Matbench Discovery to test the performance of UMLPs.
Folowing metrics are reported in the benchmark:
CPS (Combined Performance Score): a metric that weights F1,RMSD, and .
F1: harmonic mean of precision and recall, used to evaluate the performance of classification models.
DAF (Discovery Accuracy Factor): a metric that measures the accuracy of a model’s predictions in a discovery task.
Prec: precision of classifying thermodynamic stability.
Acc: accuracy of classifying thermodynamic stability.
MAE: average absolute error in convex hull distances.
RMSD: a measure of the average deviation of predicted structures from reference structures after relxation.
: error in the phonon mode contribution to thermal conductivity .
- Deng, B., Zhong, P., Jun, K., Riebesell, J., Han, K., Bartel, C. J., & Ceder, G. (2023). CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nature Machine Intelligence, 5(9), 1031–1041. 10.1038/s42256-023-00716-3
- Barroso-Luque, L., Shuaibi, M., Fu, X., Wood, B. M., Dzamba, M., Gao, M., Rizvi, A., Zitnick, C. L., & Ulissi, Z. W. (2024). Open Materials 2024 (OMat24) Inorganic Materials Dataset and Models. arXiv. 10.48550/ARXIV.2410.12771
- Schmidt, J., Cerqueira, T. F. T., Romero, A. H., Loew, A., Jäger, F., Wang, H.-C., Botti, S., & Marques, M. A. L. (2024). Improving machine-learning models in materials science through large datasets. Materials Today Physics, 48, 101560. 10.1016/j.mtphys.2024.101560
- Jacobs, R., Morgan, D., Attarian, S., Meng, J., Shen, C., Wu, Z., Xie, C. Y., Yang, J. H., Artrith, N., Blaiszik, B., Ceder, G., Choudhary, K., Csanyi, G., Cubuk, E. D., Deng, B., Drautz, R., Fu, X., Godwin, J., Honavar, V., … Wood, B. M. (2025). A practical guide to machine learning interatomic potentials – Status and future. Current Opinion in Solid State and Materials Science, 35, 101214. 10.1016/j.cossms.2025.101214