Codes
Computational Codes for High-Throughput Simulations¶
Here we will discuss the computational codes used in high-throughput (HT) simulations, focusing on density functional theory (DFT), molecular dynamics, and Monte Carlo methods. These codes are essential for calculating the properties of materials in a high-throughput manner, enabling the exploration of large materials databases and the prediction of new materials with desired properties.
Density Functional Theory¶
In HT, DFT is mainly used for:
Calculating ground-state properties: total energy, electronic band structure, density of states, magnetic properties, elastic constants, etc.
Screening for thermodynamically stable materials by comparing energies.
Predicting properties for various materials.
Popular DFT codes include:
| Code | Licencing | Feature |
|---|---|---|
| VASP | Shared-source commercial | Known for accuracy and efficiency, especially for periodic systems. Suitable for transition metals and heavy elements. Used in databases like the Materials Project. |
| Quantum ESPRESSO | Open-source | Flexible and scalable suite for electronic structure calculations and materials modeling. Suitable for a wide range of systems and properties. |
| CASTEP | Shared-source commercial/academic | Powerful for computing spectroscopy properties. Used in the Materials Studio software suite. |
| GPAW | Open-source | Python-based DFT code. Works well with ASE and uses the PAW method for pseudopotentials. |
| Gaussian | Commercial | Widely used for quantum chemistry calculations, including DFT. Known for its accuracy and extensive range of methods. |
Molecular Dynamics¶
Recall that force fields are used for studying time-dependent phenomena, thermal properties, and materials behavior under realistic conditions.
Calculating finite-temperature properties: thermal expansion, heat capacity, thermal conductivity.
Simulating diffusion, phase transitions, and materials processing.
Popular force field codes include:
| Code | Licencing | Feature |
|---|---|---|
| LAMMPS | Open-source | Highly flexible and scalable molecular dynamics code. Suitable for a wide range of materials and simulation types. |
| GROMACS | Open-source | Specialized for biomolecular simulations but also used for materials modeling. Known for its performance and efficiency. |
For Monte Carlo, the codes are usually developed in-house or adapted from existing libraries due to the specific nature of the simulations.
Performance and Reproducibility¶
These codes were written in Fortran or Python calling Fortran/C++ Libraries for high performance. They are usually run on high-performance computing (HPC) clusters due to their high computational cost. For such purpose, they are optimized for parallel computing using MPI (Message Passing Interface) or OpenMP (Open Multi-Processing) to distribute the workload across multiple processors.
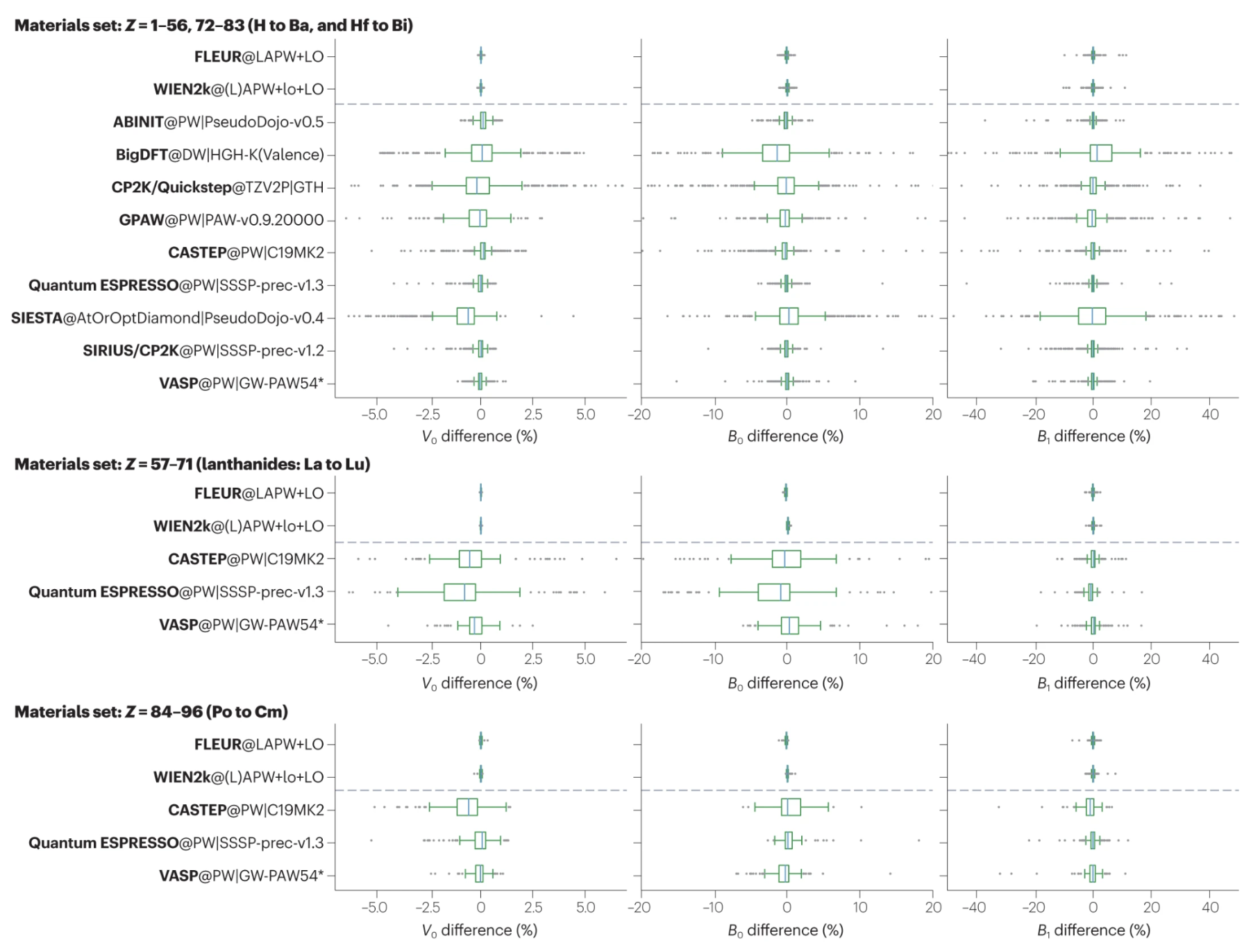
Verification of the precision of DFT codes. Reproduced from Bosoni et al..
The codes are extremely complicated, especially for DFT codes, which requires a deep understanding of DFT theory and computational methods to use effectively. The development of these codes usually needs collaborations between different research group. Most of the DFT codes can show consistent results ( value), which is important for HT calculations.
However, be careful when publishing results for comparison between different codes, as it might violate the license agreement (especially for commercial softwares).
Workflow Management Systems¶
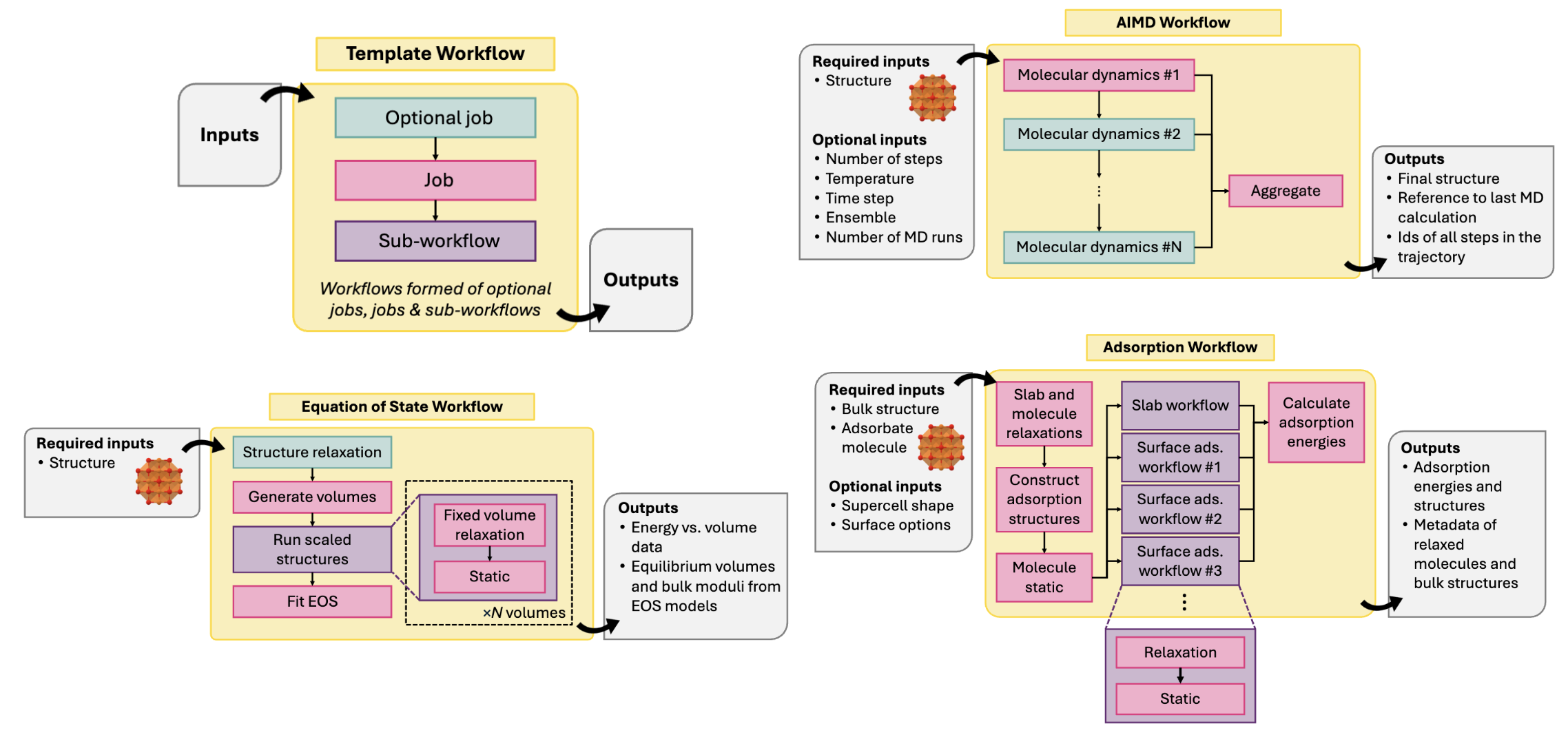
Some workflows from Atomate 2 for high-throughput calculations. Reproduced from Ganose et al..
Workflow management systems are essential for coordinating and automating the various steps in a high-throughput simulation workflow. These systems help manage the complexity of running large numbers of simulations. They can handle tasks such as setting up simulations, running calculations, and analyzing results, allowing researchers to focus on the scientific aspects of their work.
AiiDA (Automated Interactive Infrastructure and Database for Computational Science): A more general-purpose workflow management system designed for high-throughput computational science, with a strong focus on provenance tracking.
FireWorks: A workflow management system specifically designed for high-throughput calculations, often used in conjunction with pymatgen.
Custodian: A Python package that provides error detection and handling for computational workflows, ensuring that simulations run smoothly and reliably.
Atomate 2: A high-level interface for setting up, running, and analyzing high-throughput calculations using various codes.
- Lejaeghere, K., Bihlmayer, G., Björkman, T., Blaha, P., Blügel, S., Blum, V., Caliste, D., Castelli, I. E., Clark, S. J., Dal Corso, A., de Gironcoli, S., Deutsch, T., Dewhurst, J. K., Di Marco, I., Draxl, C., Dułak, M., Eriksson, O., Flores-Livas, J. A., Garrity, K. F., … Cottenier, S. (2016). Reproducibility in density functional theory calculations of solids. Science, 351(6280). 10.1126/science.aad3000
- Bosoni, E., Beal, L., Bercx, M., Blaha, P., Blügel, S., Bröder, J., Callsen, M., Cottenier, S., Degomme, A., Dikan, V., Eimre, K., Flage-Larsen, E., Fornari, M., Garcia, A., Genovese, L., Giantomassi, M., Huber, S. P., Janssen, H., Kastlunger, G., … Pizzi, G. (2023). How to verify the precision of density-functional-theory implementations via reproducible and universal workflows. Nature Reviews Physics, 6(1), 45–58. 10.1038/s42254-023-00655-3
- Ganose, A., Sahasrabuddhe, H., Asta, M., Beck, K., Biswas, T., Bonkowski, A., Bustamante, J., Chen, X., Chiang, Y., Chrzan, D., Clary, J., Cohen, O., Ertural, C., Gallant, M., George, J., Gerits, S., Goodall, R., Guha, R., Hautier, G., … Jain, A. (2025). Atomate2: Modular workflows for materials science. 10.26434/chemrxiv-2025-tcr5h